監修:岐阜大学医学部附属病院 医学教育開発研究センター 鈴木 康之
ムコ多糖とは長い鎖状の高分子で、様々な種類があり、全身の臓器や組織で重要な働きをしています(関節のコンドロイチンもムコ多糖の一種です)。このムコ多糖を分解する酵素の遺伝子に異常があり、体内にムコ多糖が蓄積すると「ムコ多糖症」を発病します。ムコ多糖が正常に存在し機能しないと、様々な症状・障害が現れ、場合によっては命を脅かすこともあります。ムコ多糖は多くの酵素が働いて分解されますので、どれか一つの酵素に異常があると、違うタイプの「ムコ多糖症」を発病します(表)。ムコ多糖を分解する酵素はいずれもライソゾームの中にあるため、ムコ多糖症も「ライソゾーム病」に分類されます。遺伝子の異常は生まれつきのもの(先天性)であり、生後間もなくから 徐々に発病する場合がほとんどです。ムコ多糖症には発見された順にローマ数字の番号で病型分類されています(欠番もあります)。また最初に発見した医師(研究者)の名前を冠した別名もついています。
表 ムコ多糖症(MPS)の分類と診断基準(折居忠夫:ムコ多糖症UPDATEより引用、一部改変)
| 病型 | 特異顔貌 | 知的障害 | 角膜混濁 | 骨の障害 | 関節障害 | 症状完成 | 尿中ムコ多糖 | 酵素異常 |
| MPS IH (ハーラー病) | 5 | 5 | 3 | 3 | 4 | 幼児 | DS、HS | α‐L‐イズロニダーゼ |
| MPS IHS (ハーラー/シャイエ病) | 3 | 2 | 2 | 2 | 3 | 幼児 | 〃 | 〃 |
| MPS IS (シャイエ病) | 2 | 0~1 | 2 | 2 | 3 | 学童 | 〃 | 〃 |
| MPS II (ハンター病、重症型) | 4 | 5 | 0~1 | 3 | 3 | 幼児 | 〃 | イズロン酸スルファターゼ |
| MPS II (ハンター病、軽症型) | 2 | 0~1 | 0 | 2 | 2 | 学童 | 〃 | 〃 |
| MPS ⅢA (サンフィリッポ病A) | 2 | 5 | 0 | 2 | 2 | 学童 | HS | ヘパランN‐スルファターゼ |
| MPS ⅢB (サンフィリッポ病B) | 2 | 5 | 0 | 2 | 2 | 学童 | 〃 | α‐N‐アセチルグルコサミニダーゼ |
| MPS ⅢC (サンフィリッポ病C) | 2 | 5 | 0 | 2 | 2 | 学童 | 〃 | *1 |
| MPS ⅢD (サンフィリッポ病D) | 2 | 5 | 0 | 2 | 2 | 学童 | 〃 | *2 |
| MPS ⅣA (モルキオ病) | 0~1 | 0 | 1~4 | 2~5 | 2~5 | 幼児 | KS-C6S | *3 |
| MPS IVB | 1 | 0 | 1 | 2 | 2 | 学童 | KS | β―ガラクトシダーゼ |
| MPS VI(マルトー・ラミー病) | 1~2 | 0 | 1~2 | 2~4 | 3~4 | 幼児 | DS | *4 |
| MPS VII (スライ病) | 2 | 3 | 1 | 1~3 | 3 | 幼児 | DS,HS,CS | β-グルクロニダーゼ |
| MPS IX | 0 | 0 | 0 | 0 | 1 | 学童 | HA | ヒアルロニダーゼ |
0,1,2,3,4,5は症状の強さを表す(0なし 5非常に強い)
DS:デルマタン硫酸、HS:ヘパラン硫酸、KS-C6S:ケラタン硫酸-コンドロイチン6硫酸複合体、CS:コンドロイチン硫酸、HA:ヒアルロン酸
*1: アセチルCoA:α-グルコサミニドN-アセチルトランスフェラーゼ
*2: N-アセチルグルコサミン-6-スルファターゼ
*3: N-アセチルガラクトサミン-6-スルフェイト スルファターゼ
*4: N‐アセチルガラクトサミン‐4‐スルファターゼ
1. ムコ多糖症はどのようにして発病するか?
人間の体は約37兆個の細胞からできています。そして、一つ一つの細胞の中には、遺伝情報を含む染色体(核)、エネルギーを作り出すミトコンドリア、タンパク質を作る小胞体、そして細胞内の様々な物質の分解・リサイクルを担っているライソゾーム(リソソーム)などが含まれています。ライソゾームの中には高分子のタンパク質、脂肪、ムコ糖などを分解する酵素(タンパク質)が多く含まれています。酵素の設計図は染色体の中にある遺伝子に書き込まれていますので、遺伝子に異常があると酵素が正常に働かなくなります。例えば、あるムコ多糖を分解する酵素の遺伝子に異常があると、そのムコ多糖が細胞内や組織内に溜まってきて、結果的に臓器の機能が悪くなります。これがムコ多糖症の本態です。ライソゾームには50種類以上もの酵素があり、それぞれ特定のタンパク質、脂肪、ムコ多糖などを分解します。そのうち11種類の酵素がムコ多糖症の発病に関係していることが知られています。
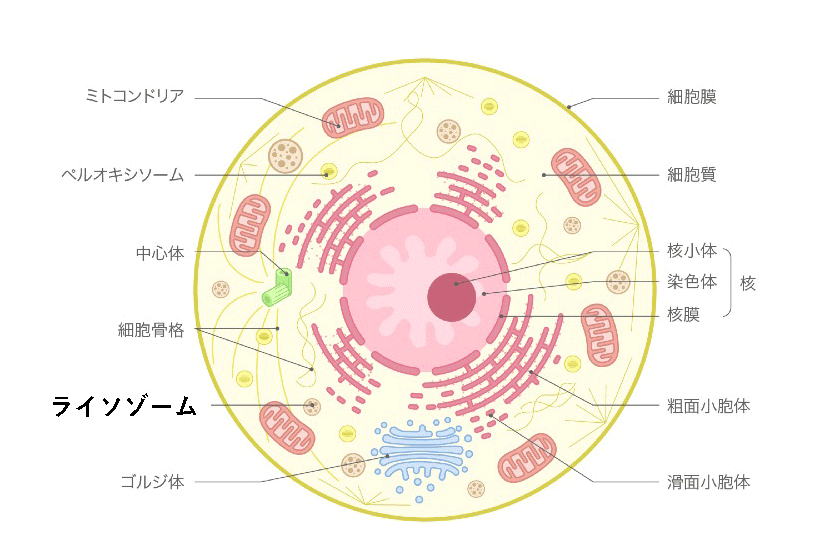
ムコ多糖症は、ムコ多糖を分解する酵素が生まれつき欠損しています。そして欠損している酵素の種類によって病気のタイプ(型)が色々あり、症状も少しずつ違います。ムコ多糖症では全身の細胞のライソゾームにムコ多糖が蓄積し、細胞は膨れ上がり、その結果、色々な臓器が肥大し機能障害を起こしていきます。赤ちゃんのときは一見健康に見えますが、年齢が進むにつれて次第に症状が現れます。症状の現れ方は、酵素の種類、障害の程度によって個人差があり、乳幼児期から症状が早く進む重症型から、比較的ゆっくりと発病し進行する軽症型まで千差万別です。
ムコ多糖症は遺伝性の病気で、II型だけがX連鎖潜性(劣性)遺伝、そのほかの型は常染色体性潜性(劣性)遺伝という遺伝の仕方をします。常染色体性潜性(劣性)遺伝では、両親が二人とも保因者であるときに、1/4の確率で病気の子供(男女差なし)が産まれます。保因者とは病気の遺伝子を半分だけ持つ人で、原則的に発病しません。片親だけが保因者の場合、一般的に病気の子どもは産まれませんが、まれに精子や卵子の中で突然変異がおきて、子どもが発病することがあります。X連鎖潜性(劣性)遺伝では、母親が保因者のことが多く、男の子は1/2の確率で発病します。女の子は病気にはなりませんが、1/2の確率で母親と同じ保因者になります。
2. 各病型の特徴
a. ムコ多糖症I型(ハーラー病、ハーラー/シャイエ病、シャイエ病)
α-L-イズロニダーゼという酵素の欠損により発病します。ムコ多糖症の中で最初に発見されたことからI型と命名され、発見した医師の名前をとって、重症型はハーラー病、軽症型はシャイエ病、中間型はハーラー/シャイエ病、とも呼ばれます。欧米ではI型患者が最も多いのですが、日本ではII型よりも少なく、ムコ多糖症全体の約15%程度です。
ハーラー病は最も重症で、乳児期から臍ヘルニア(でべそ)・そけいヘルニア(脱腸)、濃い蒙古斑が高率に認められ、肝臓・脾臓が大きく、骨の変形、特徴的な顔つき、関節の拘縮、大きな舌などに気づかれます。乳児期の体格は大きめで骨太ですが、その後、成長は停滞し、最終身長は100cm程度です。知的理解の発達も遅れ、2〜4歳以降はそれまで出来ていたことが出来なくなります(退行)。呼吸器の感染や中耳炎を繰り返し、難聴が進行します。目では角膜混濁や緑内障が起こります。特徴的な体型となり、頭蓋骨が大きく肥厚し、全身関節の拘縮、背骨が変形して背中が飛び出したようになります(突背)。
シャイエ病は軽症型で、幼児期以降に関節拘縮、心臓弁膜症、角膜混濁、肝臓・脾臓の腫大、特徴的な顔貌、関節の変形・拘縮、呼吸障害・睡眠時無呼吸などが徐々に生じますが、知的能力は正常で、成人して仕事に就いている方も多くおられます。身長は130~150cmまで伸びます。角膜混濁・緑内障・網膜変性などにより視力障害が生じます。
治療法:酵素補充療法、造血幹細胞移植(骨髄移植)、対症療法
b. ムコ多糖症II型(ハンター病)
イズロン酸スルファターゼという酵素の欠損により発病します。日本人のムコ多糖症の50%以上を占め、最も多い病型です。I型同様、重症型から軽症型まで様々な重症度の患者さんがおられます。症状はI型とよく似ており、重症型では1歳頃から特徴的な顔つき、骨の変形、関節の拘縮、肝臓・脾臓の腫大などが始まり、「言葉の遅れ」が診断のきっかけになることが多いです。背中の濃い蒙古斑、臍ヘルニア、そけいヘルニア、中耳炎もよく合併します。5〜6歳ころから退行が始まり、呼吸障害や心不全が進行します。
軽症型では5歳頃から重症型と同様の症状に気づかれますが、進行は緩やかで、知的能力は正常の方が多く、成人して仕事に就いている方も多くおられます。身長も130~150cm位になります。難聴、視力障害、関節拘縮、神経障害、心不全、呼吸不全などのために生活に支障をきたします。
治療法:酵素補充療法、造血幹細胞移植(骨髄移植)、対症療法
c. ムコ多糖症III型(サンフィリポ病)
原因酵素(遺伝子)の違いからA型、B型、C型、D型に分類されますが(表参照)、症状はほぼ同じです。日本人ではII型に次いで多く、ムコ多糖症全体の約20%を占めています。他のムコ多糖症と異なり、神経症状が特徴的で、2〜6歳頃から落ち着きがない、興奮、乱暴な行動、言葉の遅れ,不眠などに気づかれます。意味のあることばが出ないままのお子さんもおられます。身体症状は軽く、多毛が比較的目立つ程度ですので、診断が遅れることが多いです。10歳代になると、睡眠障害、肝脾腫、痙攣発作が見られ、周囲との意思疎通が困難となります。
治療法:対症療法
d. ムコ多糖症IV型(モルキオ病)
原因酵素(遺伝子)の違いからA型とB型に分類されますが(表参照)、A型が多く、ムコ多糖症の約10%を占めています。骨の変形が非常に強く、重症の方は身長も100cm前後ですが、知能は障害されません。幼児期から手足の変形、背骨のゆがみ、短い首と胴、X脚、低身長に気づかれます。背骨が強く変形して扁平になるために胴が短くなります。関節の靱帯が弛緩するために、関節がぐらぐらと不安定になります。頸椎(背骨の首の部分)の形成不全などにより脊髄が圧迫され、手足の麻痺が生じることもあります。
治療法:酵素補充療法、整形外科的治療、対症療法、造血幹細胞移植
e. ムコ多糖症VI型(マルトー・ラミー病)
I型(ハーラー病)に似た身体所見を示しますが、知能は障害されません。1歳頃から大きな頭、骨の変形、関節拘縮が認められ、臍ヘルニア、そけいルニアも多く見られます。角膜混濁、肝臓・脾臓の腫大、骨の変形、心臓弁膜症などが見られます。
治療法:酵素補充療法、造血幹細胞移植,対症療法
f. ムコ多糖症VII型(Sly病)
症状は多様で、ハーラー病のような全身症状・神経症状をきたすタイプ、モルキオ病のような骨の変化が主体のタイプ、胎児期から全身がむくむタイプ(胎児水腫)などがあります。
治療法:対症療法、造血幹細胞移植、酵素補充療法(米国)
g. ムコ多糖症IX型(ヒアルロニダーゼ欠損症)
関節の腫れや痛みを主体とする病型で、世界で数例しか報告がなく、日本ではまだ患者さんが見つかっていません。
治療法:対症療法
3. 診断
ムコ多糖症を疑う症状が認められた場合、尿のムコ多糖分析を行います。ムコ多糖(デルマタン硫酸、ヘパラン硫酸、ケラタン硫酸)の排泄が多く認められたら、さらに酵素診断、場合によっては遺伝子診断を行い、診断を確定します。一部の都道府県では、新生児マススクリーニングの検査項目の中にムコ多糖症の一部が含まれ、希望者には有料で実施されています。
ムコ多糖症|型ガイドライン
http://www.japan-Isd-mhlw.jp/doc/mps1 practice-guideline 2019.pdf
ムコ多糖症II型ガイドライン
http://isimd.net/pdf/MPS2019.pdf
難病情報センター ライソゾーム病とは
https://www.nanbyou.or.jp/entry/4063

賛助会員について
皆様が支えて下さることが、患者ご家族にとって大きな励みに、大きな支えになります。
私たちの活動を援助して下さる「賛助会員」にご登録をお願いします。

正会員のご案内
事前に電話やメールでの連絡も受けておりますので、ご質問等ありましたらご連絡下さい。

ご寄付のお願い
皆様のご協力をよろしくお願いします。










